Case 46
Author: James Leong Editor: Adrian Fung
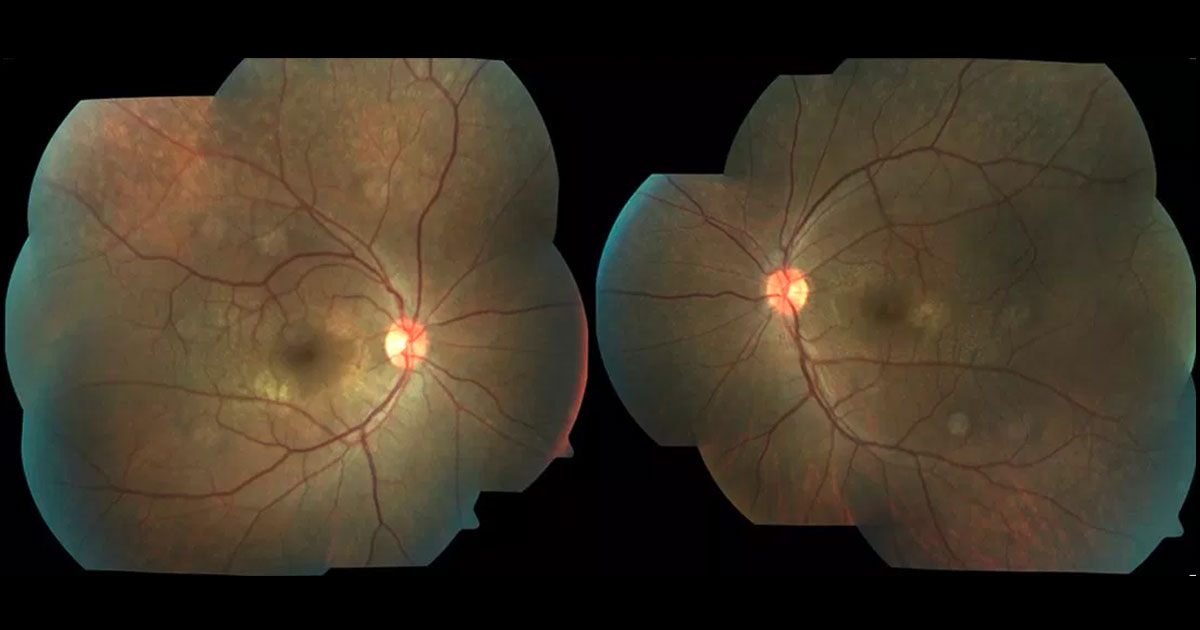
Figure 1. Colour fundus photography demonstrating perimacular flecks at both maculae.
A 35-year-old male was referred for an opinion regarding incidental bilateral macular findings.
Case history
A 35-year-old asymptomatic mild myope was referred with bilateral macular findings. The patient had a history of renal failure requiring renal transplant and immunosuppression. On examination his visual acuities were 6/9 bilaterally and intraocular pressures were normal. Both corneae were clear and there were no cataracts. Posterior segment examination demonstrated a bilateral perimacular fleck retinopathy (Figure 1 and 2).
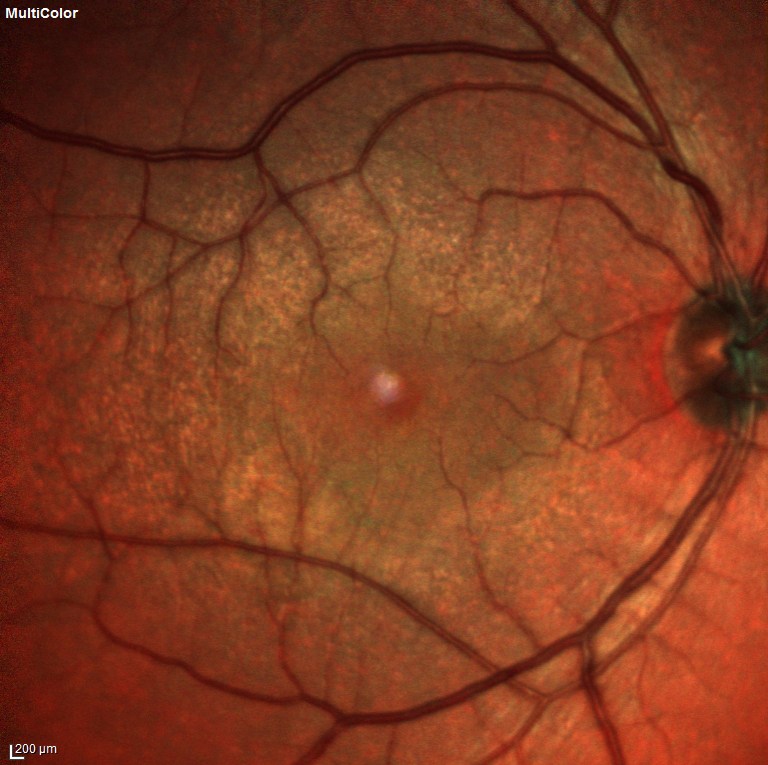
Figure 2. Multicolour image showing the flecks in more detail.
What is your diagnosis?
Click to reveal answer
Differential diagnosis
The differential diagnosis of a fleck retinopathy include a number of retinal dystrophies:
- Stargardt / Fundus Flavimaculatus (ABCA4)
- Mulitfocal Pattern Dystrophy (peripherin/RDSgene)
- Dominent drusen
- Fundus Albipunctatus
- Benign Fleck Dystrophy
- Alport Syndrome
- Cystinosis
- Hyperoxaluria
Additional history, examination and investigations
Optical Coherence Tomography through the lesion in the left eye showed it to be a hyper-reflective band, located at the inner nuclear layer (INL, Figure 2). The other retinal layers and the left OCT were normal. Fundus autofluorescence (Figure 3), fluorescein angiography (Figure 4) and Humphrey Visual Field testing (Figure 5) were normal in both eyes. However, near-infrared reflectance demonstrated a dark, wedge-shaped lesion corresponding to the white lesion on clinical examination.

Figure 3. Anterior segment photograph demonstrating anterior lenticonus.
Figure 4. Pentacam corneal topography confirms anterior lenticonus.
Figure 5. Optical Coherence Tomography demonstrates marked thinning of the temporal retina.
DIAGNOSIS
The constellation of findings including anterior lenticonus, perimacular fleck retinopathy and temporal thinning on OCT imaging is consistent with Alport syndrome.
Clinical course
Subsequent genetic testing confirmed Alport syndrome. Genetic counselling was provided for the patient and his family. Audiometry was arranged to further assess his hearing loss. Annual reviews were arranged to monitor the patients progress including for the development of cataracts and macular holes.
Discussion
Alport syndrome is an inherited disorder characterized by progressive renal failure, sensorineural hearing loss and ocular manifestations.(1) It is typically an X-linked condition caused by mutations in the COL4A5 gene. This gene codes for Collagen IV which is a basement membrane protein seen in the kidneys (glomerular basement membrane), ears (cochlear) and eyes (Descemet’s Membrane, Bowman’s membrane, lens capsule, internal limiting membrane and Bruch’s membrane).
The characteristic ocular manifestations of Alport syndrome include corneal opacities, anterior lenticonus, central or peripheral fleck retinopathy and temporal macular thinning.(1,2,3) The central retinopathy can be seen as perimacular scattered whitish–yellow dots and flecks. These ocular manifestations of Alport syndrome typically do not impair vision. Rarely the patients may develop metamorphopsia or reduced visual acuity due to the development of lamellar or full thickness macular holes.(4)
The ocular findings are particularly useful in assisting the diagnosis of Alport syndrome. This is particularly important because detection and early treatment with angiotensin-converting enzyme inhibitors can slow progression of the renal impairment.(5) An established diagnosis also has implications regarding the risk of the disease in other family members.
TAKE HOME POINTS
- Alport Syndrome is an inherited condition characterised by ocular abnormalities, progressive renal failure and hearing loss.
- Ocular findings include corneal opacities, anterior lenticonus, fleck retinopathies and temporal macular thinning.
REFERENCES